









Fluorescein(FITC) Tunel Cell Apoptosis Detection Kit(50T)
%20Tunel%20Cell%20Apoptosis%20Detection%20Kit-550x550w.jpeg "Fluorescein(FITC) Tunel Cell Apoptosis Detection Kit(50T)")
- Stock: In Stock
- Model: 0442

Product Description/Introduction
The breakage of chromosomal DNA in apoptosis is a gradual process. Chromosomal DNA is first degraded into large fragments of 50-300 KB by endogenous nuclease hydrolase, and then about 30% of chromosomal DNA is randomly cut between nucleosome units under the action of Ca2+ and Mg2+ dependent nucleic acid endonuclease to form 180-200 bp nucleosome DNA polymers. Therefore, in late apoptosis, DNA is degraded into 180-200 bp fragments, and a large number of 3'-OH terminals are exposed on the broken genomic DNA. Terminal deoxyribonucleotidyl transferase (TDT) is a template independent DNA polymerase, which can catalyze the binding of deoxynucleotides to the 3'-OH terminal of broken DNA molecules. Therefore, TUNEL (TDT mediated dUTP nick end labeling) cell apoptosis detection kit can be used to detect the breakage of nuclear DNA in tissue cells during the late apoptosis. It is based on the incorporation of fluorescein-labelled dUTP (FITC-12-dUTP) at the exposed 3´-OH ends of genomic DNA breaks by TdT enzymes, which can be detected by fluorescence microscopy or flow cytometry (FITC excitation 495 nm, emission 521 nm). This kit is suitable for apoptosis detection of paraffin tissue sections, frozen tissue sections, cell crawling, cell smears.
Storage and Shipping Conditions
Ship with wet ice; This kit store at -20°C, FITC-12-dUTP Labeling Mix store at -20°C protect from light and valid for 12 months.
Product Components
Component | 0442 | ||
Recombinant TdT Enzyme | 50 µL | ||
FITC-12-dUTP Labeling Mix | 250 µL | ||
Equilibration Buffer | 5×1 mL | ||
Proteinase K (200 µg/mL) | 1 mL | ||
Manual | 1 pc | ||
Product Preparation
1. PBS phosphate buffer (recommended G0002 or G4202).
2. Fixative solution: 4% paraformaldehyde dissolved in PBS or other buffer systems, pH 7.4 (recommended G1101).
3. Membrane breaking liquid:0.1%-0.5% Triton X-100 (recommended G1204).
4. If you need to dye the nucleus, you need to bring your own DAPI (2 µg/mL)or PI (1 µg/mL) (recommended G1012,G1021).
5. For positive control experiments, DNase I is required (recommended G3342).
6. If using a flow cytometer, bring your own PI staining solution (recommended G1021)and RNase A (DNase free).
7. For your safety and health, please wear safety glasses, gloves, or protective clothing.
Product Protocol/Procedures
1. Sample Preparation
A. Paraffin Tissue Sections
1) Soak paraffin tissue sections in BioDewax and Clear Solution (G1128) for 5-10 min at room temperature and repeat 3 times; Then in anhydrous ethanol for 5 min and repeated twice; Finally in gradient ethanol (85%, 75%, and double distilled water) once for 5 min each time.
2) Wash slices gently with PBS and remove the excess liquid around the sample. Use a histochemical pen to draw a small circle 2-3 mm apart along the peripheral contour of the tissue to facilitate downstream permeability treatment and balance labelling. During the experiment, do not let the sample dry, and put the treated sample in the wet box to keep the sample moist.
3) Prepare Proteinase K working solution: dilute Proteinase K (200 µg/ml) stock solution with PBS at a ratio of 1:9 to a final concentration of 20 μg/mL.
4) Add 100 μL of Proteinase K working solution to each sample, completely cover the tissue and incubate at 37 ℃ for 20 min.
Note: Proteinase K contributes to the permeation of staining reagents in the subsequent steps of the tissue and cells. Incubate for too long or too short will affect the efficiency of the subsequent labelling. For better results, the incubation time of Proteinase K can be optimized.
5) Soak and clean the sample with PBS solution for 3 times, each time for 5 min (Proteinase K needs to be cleaned, otherwise it will interfere with the subsequent marking reaction). keep the sample moist in the wet box after treatment.
6) (optional steps) remove the excess liquid from the sample, add an appropriate amount of membrane breaking liquid to the tissue, fully infiltrate the tissue, and treat at room temperature for 20 min; After the membrane breaking treatment is completed, wash the sample three times with PBS solution for 5 min each time; keep the sample moist after treatment in a wet box.
B. Frozen Tissue Sections
1) Immerse the slides in 4% paraformaldehyde solution (dissolved in PBS) for fixation, and incubate at room temperature for 10-15 min.
2) Remove the film from the fixation solution and let it dry naturally in a fume hood.
3) Wash the slide in pure water or PBS to remove residual fixation solution from the slide.
4) Use a histochemical pen to draw a small circle 2-3 mm apart along the peripheral contour of the tissue to facilitate downstream permeability treatment and balance labelling. During the experiment, do not let the sample dry, and put the treated sample in the wet box to keep moist.
5) Prepare Proteinase K working solution: dilute Proteinase K (200 µg/ml) stock solution with PBS at a ratio of 1:9 to a final concentration of 20 μg/mL.
6) Add 100 μL of Proteinase K working solution to each sample, completely cover the tissue and incubate at 37 ℃ for 10 min.
Note: Proteinase K contributes to the permeation of staining reagents in the subsequent steps of the tissue and cells. Incubate for too long or too short will affect the efficiency of the subsequent labelling. For better results, the incubation time of Proteinase K can be optimized.
7) Rinse the sample with PBS solution for 2-3 times to remove the excess liquid (Proteinase K needs to be cleaned, otherwise it will interfere with the subsequent marking reaction). keep the sample moist in the wet box after treatment.
8) (optional steps) add an appropriate amount of membrane breaking liquid to the tissue, fully infiltrate the tissue, and treat it at room temperature for 20 min. After completion of the membrane breaking treatment, wash the sample with PBS solution to remove the excess liquid. keep the sample moist after treatment in a wet box.
C. Cell Crawling
1) Adherent cells are cultured on lab-Tek (Chamber Slides). After apoptosis induction treatment, the slides are gently rinsed twice with PBS.
2) Add an appropriate amount of 4% paraformaldehyde solution (dissolved in PBS) to each slide chamber for fixation, and incubate at room temperature for 20 min.
3) Remove the fixation solution and wash three times with PBS, each time for 5 min.
4) Immerse samples in the membrane breaking solution and incubate at room temperature for 5 min for permeabilisation.
5) Immerse and clean the sample in an open beaker containing PBS solution for 2-3 times.
6) Gently remove excess liquid and carefully drain the liquid around the sample on the slide with filter paper. Keep the sample moist after treatment in a wet box.
D. Cell Smear
1) Resuspend cells in PBS at a concentration of approximately 2×107 cells/mL, pipette 50-100 μL of cell suspension onto an anti-defossing slide, and gently spread the cell suspension using a clean slide.
2) Immerse the slides in a staining jar filled with 4% paraformaldehyde freshly formulated in PBS, fix the cells, and leave at 4 °C for 25 min.
3) Immerse the slide in PBS, place it at room temperature for 5 min, and repeat once.
4) Gently remove the excess liquid, and carefully drain the excess liquid around the sample on the slide with filter paper, draw a small circle along the outer contour of the cell with a histochemical pen to facilitate downstream permeability processing and balance labeling operations, and do not allow the sample to dry during the experiment.
5) Immerse samples in the membrane breaking solution and incubate at room temperature for 5 min for permeabilisation.
6) Immerse and clean the sample in an open beaker containing PBS solution for 2-3 times.
7) Gently remove excess liquid and carefully drain the liquid around the sample on the slide with filter paper. Keep the sample moist after treatment in a wet box.
2. DNase I Treatment Positive Control Experiment (Optional Steps)
After the sample permeability treatment, treat the sample with DNase I (recommended G3342) to prepare the positive control.
1) Add 100 μL of 1× DNase I buffer (preparation method: mix well 10 μL of 10× DNase I buffer and 90 μL of deionized water ) to the permeable sample and incubate at room temperature for 5 min.
2) Gently remove excess liquid and add 100 μL of working solution containing DNase I (20 U/ml) (preparation method: mix well 10 μL of 10× DNase I buffer, 2 μL of DNase I and 88 μL of deionized water), incubate at room temperature for 10 min.
3) Gently remove the excess liquid, and thoroughly wash the slide 3-4 times in the staining tank with PBS;
(Note: the positive control slide must use a separate staining cylinder, otherwise the residual DNase I on the positive control slide may introduce a high background on the experimental slide).
3. Marking and Testing
1) Equilibration: Add 50 μL of Equilibration Buffer per sample to cover the sample area and incubate at room temperature for 10 min.
2) Labeling solution preparation: thaw FITC-12-dUTP Labeling Mix and Equilibration Buffer on ice and mix sufficient TdT incubation buffer according to the ratio of Recombinant TdT enzyme: FITC-12-dUTP Labeling Mix: Equilibration Buffer=1 μL: 5 μL: 50 μL (1: 5: 50). The volume of reagents used in specific experiments can be adjusted in an appropriate equal proportion according to the size of the slide.
3) Negative control system: Prepare a control TdT incubation buffer without Recombinant TdT enzyme and replace it with ddH2O.
4) Labeling: remove the equilibration Buffer and then add 56 μL of TdT incubation buffer to each tissue sample and incubate at 37 °C for 1 h; Note that the slides should not be dried and protected from light.
5) Immediately wash the tissue samples with PBS four times for 5 min each.
6) Gently wipe off the PBS solution around the sample with filter paper.
7) Nuclear staining: Stain the samples in a staining vat and immerse the slides with PI solution or DAPI solution (freshly formulated and diluted with PBS) in the dark at room temperature for 8 min.
8) Sealing: After staining, wash the sample three times with PBS for 5 min each, then gently remove the excess liquid and sealing the slide with the anti-fluorescence quenching sealer (recommended G1401)).
9) Microscopy: Immediately analyze the sample under a fluorescence microscope, the slides are carefully protected from light, PI/DAPI can stain both apoptotic and not apoptotic cells red/blue, green fluorescence localized by FITC-12-dUTP incorporation only in the nuclei of apoptotic cells.
4. Suspension Cells are Detected Using Flow Cytometry
1) The cells to be detected are washed twice with PBS, centrifuge (500 x g) at 4°C and resuspended in 500 μL of PBS.
2) Fixation: Add 5 mL of 1% paraformaldehyde solution prepared with PBS to the sample, fix the cells, and leave on ice for 20 min.
3) Cells are centrifuged at 4 °C, 300 x g for 10 min, supernatanted and resuspended twice with 5 mL of PBS, and finally resuspended with 500 μL of PBS.
4) Permeability: Add 5 mL of pre-cooled 70% ethanol on ice to the sample and incubate at -20°C for 4 h to permeabilise the cells.
(Note: Cells can also be permeated with membrane breaking solution at room temperature for 5 min)
5) Cells are resuspended with 5 mL of PBS after centrifugation at 300 x g for 10 min, and resuspended with 1 mL of PBS after centrifugation again.
6) Equilibration: Transfer approximately 2×106 cells to a 1.5 mL microcentrifuge tube, centrifuge at 300 x g for 10 min, discard the supernatant and resuspend with 80 μL of Equilibration Buffer, incubate at room temperature for 5 min.
7) Labeling solution preparation: thaw FITC-12-dUTP Labeling Mix and Equilibration Buffer on ice and mix sufficient TdT incubation buffer according to the ratio of Recombinant TdT enzyme: FITC-12-dUTP Labeling Mix: Equilibration Buffer=1 μL: 5 μL: 50 μL (1: 5: 50) for all experiments and optional positive control reactions.
8) Labeling: Cells are centrifuged at 300 x g for 10 min, discard the supernatant and resuspend the precipitate in 56 μL of TdT incubation buffer, incubate at 37°C for 1 h, protect from light. Gently resuspend cells with a micropipette every 15 min.
9) After the reaction is complete, add 1 mL of 20 mM EDTA to terminate the reaction and mix gently with a micropipette.
10) Centrifuge at 300 x g for 10 min, discard the supernatant and resuspend the precipitate in 1 mL of membrane breaking solution containing 5 mg/mL BSA, repeat wash twice.
11) Nuclear staining: Centrifuge at 300 x g for 10 min, discard the supernatant and resuspend the cell precipitate in 0.5 mL of PI solution containing 250 μg of RNase A without DNAase, incubate the cells in the dark at room temperature for 30 min.
12) Hands-on detection: Flow cytometry analyzes cells, PI can dye both apoptotic and unpoptoptotic cells red/blue, and only in the apoptosis nucleus does FITC-12-dUTP incorporate and locate the green fluorescence.
5. Experimental Process Diagram
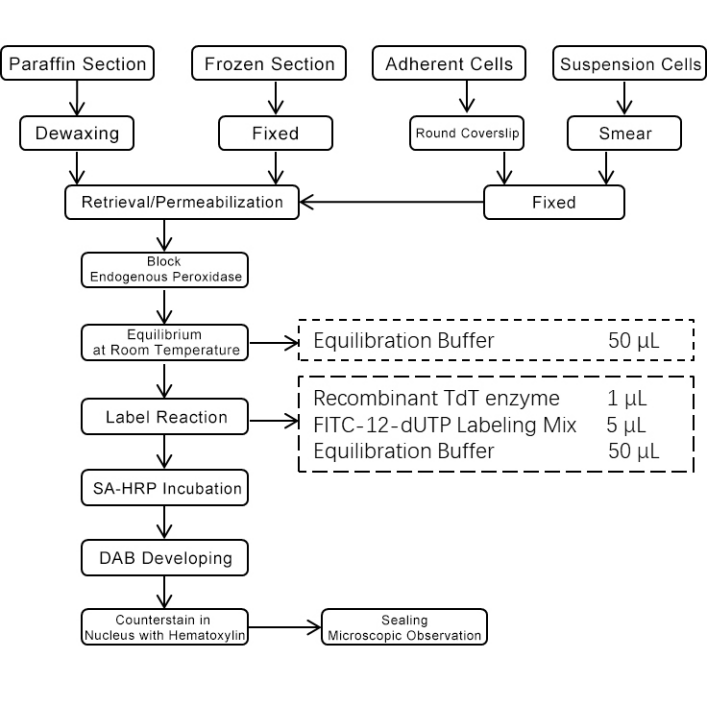
Note
For your safety and health, please wear safety glasses, gloves, or protective clothing.
| Thumb | File information |
| G1501-Fluorescein FITC Tunel Cell Apoptosis Detection.pdf File Size283.69KB Downloaded: 113 |
1-250x250.jpeg "2 × Fast SYBR Green qPCR Master Mix (High ROX) 1mL")
1-250x250.jpeg "2 × Fast SYBR Green qPCR Master Mix (Low ROX) 1mL")
 1mL")
 1mL")
 1mL")
")

")